Development of drugs remain an incredibly important factor in human existence. While the world seems to face shortage of medicines because of increasingly new diseases, scientists in pharmaceutical industries and academic researchers are busy looking for new strategies of discovering and developing either entirely new drugs (new chemical entities) or new drugs from existing compounds.
This paper reviews how changing the route of administration of an already approved drug can be used to incrementally optimize an old drug, giving benefits like increased solubility and permeability, by-passing first pass effect, decreased cost, increased patient compliance to treatment, etc. Additionally, this paper summarizes the 505(b)(2) program which is the new drug application pathway for the approval of such new drug.
The approach employed in this review was studying information from literatures, company and government websites, and examples from official books.
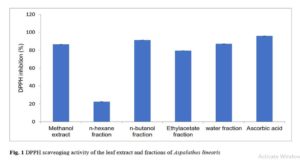
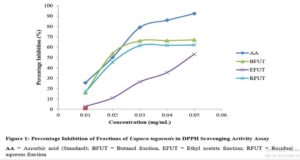
Results show that although changing a drug’s route of administration is possible and encouraged, there are important tests for filing any modification in routes of administration, either clinical or non-clinical. Also presented are examples of drug products that have seen such changes.
In conclusion, incrementally modifying drugs through changing route of administration has been seen to potentiate the use and value of important medicines, and their importance in a large population of patients cannot be overemphasized.