1.0 INTRODUCTION
The incrementally modified drugs refer to medicines that have similar active ingredients and efficacy to the innovator drugs (the Listed Drug) but have varied properties, manufacturing processes and other requirements to deliver therapeutic advantages yet maintain safety and effectiveness. Original drugs in this case can be existing or old drugs that have been approved and are in use, or a drug that was not successful, or a drug that was withdrawn for various reasons. The Korean Ministry of Food and Drug Safety recognizes those products as incrementally modified drugs which are backed up by scientific proofs, with more flexible development requirements than innovator brands regarding quality, safety, efficacy, and ease of use. The clinical development timeline of the incrementally modified drugs is shorter than those of new chemical entities, risk of product failure is lower, and trials/application costs are also lesser. Making changes to an existing drug by altering its route of administration can be beneficial in increasing patient compliance to therapy, avoiding enzyme metabolism (CytP450 activities), by-passing risk of degradation by gastro-intestinal tract enzymes, improving solubility and permeability of some drugs, etc. Some drugs have been modified by changing their administration routes for instance, the Daehwa Pharmaceutical innovative anti-tumor drug, Liporaxel® Solution transformed paclitaxel injection (an existing anticancer agent) into an orally administered anti-tumor drug. Additionally, the prokinetic drug Mosapride citrate was modified into a once-a-day controlled release product called GastiinCR® made by Korea United Pharm and approved in 2016 (Lee Hye-seon 2017). Controlled or Extended Release as a term means that the drug product slowly dissolves in the system, and this activity reduces the drug dosage and potential adverse effects.
Owing to the investment costs associated with developing new chemical entities, pharmaceutical research and development units have begun to create innovative options for repositioning or redeveloping existing drugs. Some innovation-driven companies expand their product value thriving on theories like activities to lower study sessions and development risk, actions to reduce development costs, and those to enhance potential for progress (Schuhmacher et al, 2016).
Benefits of Incrementally Modified Drugs
In a review by Frasier Institute (Globerman and Lybecker, 2014), innovations from pharmaceutical industries result in drugs and top-notch therapies that improve quality of life and decrease mortality, while equally enriching the innovator company and other stakeholders. Advances and improvements on already approved or existing drugs can be termed incremental with positive feedbacks like follow-on products or hybrid products or biosimilar products (in case of biologics). With incrementally modified drugs, patients can select from a wide variety of brands within a single therapeutic class, depending on need, access, and affordability. This gives physicians options for fostering targeted therapy. This option therefore enhances patient compliance to treatment and paves way for better health outcomes (Globerman and Lybecker, 2014). Further, reflecting on a meta-analysis in 2018 (Komadja et al, 2018), researchers emphasized accruable benefits of incrementally combined new drugs for chronic heart failure (combinations of beta blocker + mineralocorticoid receptor antagonist + angiotensin receptor neprilysin-inhibitor and those of mineralocorticoid receptor antagonist + beta blocker + angiotensin-converting enzyme inhibitor+ Ivabradin showing better effectiveness in their research). These benefits include continuing decrease in mortality potential and progress in hospitalization outcomes for patients (Komadja et al, 2018). In a short note, incremental medicines provide benefits such as
- Increased efficacy from old drug
- Lowered side effects due to change of administration route, formulation, etc
- Less drug intake as a result of improved dose, frequency, and duration, hence improving patient compliance
- Modified delivery designs and systems leading to enhanced bioavailability (Mithilesh 2012).
Reformulating existing medicines can be done by
- Increasing permeability especially for biopharmaceutical class (BCS) III and IV drugs using intestinal membrane permeation enhancers, P-glycoprotein inhibitors, surfactant vehicles, ion pairing
- Enhancing solubility (BCS class II and IV drugs) through particle size reduction, solid dispersion and co-crystallization, complexation techniques
- Controlled or modified release formulations
- Polyethylene glycol chain addition to delay clearance
- Employing precision by targeted delivery and local delivery (as in transdermal, etc).
The significance of this review to elucidate the various kinds of studies (clinical and nonclinical) that can be undertaken or data to be submitted if a manufacturer desires to modify a given drug and file for a new drug application (NDA) with focus on changing its route of administration. Knowing the right drug approval pathway for submitting a new drug product application to FDA requires an understanding of the available guidelines and the types of data that are required to support the filing.
In 1984, the US Food and Drug Administration (FDA) created The Hatch-Waxman Amendments that added sections 505(b)(2) and 505(j) to the Federal Food, Drug, and Cosmetic Act (FD&C Act), making room for securing new drug approvals and Abbreviated New Drug Applications at a shorter time (ANDAs) (ANDA;505(B)(2) 2019). The publication on the US FDA’s final guidance for industry which focuses on “Determining Whether to Submit an ANDA or 505(b)(2) Application” aids potential applicants by offering guidance in establishing whether to use the 505(b)(2) or ANDA pathway (ANDA;505(B)(2) 2019). For incrementally modifying drug through changing route of administration, the 505(b)(2) pathway seem to be a better process according to US FDA Center for Drug Evaluation and Research (CDER).
1.1 Fundamentals Of 505(B)(2) NDA Program
505(b)(2) program is considered a full New Drug Application (NDA) for which the applicant product (that is the new drug of interest) must be of the same Active Pharmaceutical Ingredient (API) with the Listed Drug (LD, the reference). In this program, applications accepted are those that support submissions that contain full reports of assessments of safety and effectiveness, but where at least some of the data essential for approval comes from studies not piloted by or for the applicant and for which the sponsor/applicant of the product has not gained a right of reference (Applications Covered by Section 505(b)(2) FDA 1999).
Documents to be submitted, known as the reporting/filing requirements include quality report, previously reported nonclinical and clinical information. These information may include studies that were not conducted by the applicant, information relying on FDA’s prior reports on safety and/or efficacy from other NDAs, information where applicant lacks right of reference, and other literature references. Depending on the type of new drug being proposed, new studies to support changes – mainly new clinical studies to support differences in the product may be required (Applications Covered by Section 505(b)(2) FDA 1999).
1.2 Products Allowed Under The 505(B)(2) NDA Program
Supplementary approvals for novel dosages, formulations, and indications have long been used for incremental modifications to drug products. It has been discussed that applicant product must be that which has change(s) to a previously approved drug (Berndt, cockburn, and Grepin, 2006). These adjustments include
- Change in Route of Administration e. g intravenous to intramuscular or subcutaneous routes
- New Active Ingredients e. g different salt component, racemates, enantiomer, complex compounds, or ester of the same active ingredient
- New Chemical/Molecular entity e. g pro drug of an existing compound , active metabolite of an old and approved drug
- New Combination product e. g drug-drug combination of active substances that were individually approved previously
- New Formulation e. g excipients not allowed under generic applications (505(j))
- New Dosage Form e. g oral to transdermal, immediate release to extended release, lotion to foam
- New Strength e. g higher or lower transitions
- New Dosage Regimen e. g twice daily to once a day regimen
- Change or addition of indication
- Switch from Prescription to Over-The-Counter product (Berndt, cockburn, and Grepin, 2006).
Additionally, products allowed for this pathway are those that
- Are neither bioequivalent to, nor inferior in bioavailability to the Listed Drug (reference drug)
- Have formulation changes outside 505(j) limits
- Cannot be used for products eligible for Abbreviated New Drug Application.
Hence, drug applications not permitted under the 505(b)(2) NDA are
- Applications for products that are covered by 505(j) for generics
- Products for which the only difference is the extent to which the absorbed active ingredient is less than the Reference Listed Drugs
- Where the only difference is that the rate of which the active ingredient is absorbed is unintentionally less than the Reference Listed Drug.
1.3 Required Data for 505(b)(2) NDA
The Sanjay Seghal notes on Streamlining the Development and approval processes for 505 (b)(2) NDAs (2013) summarized required data for 505 (b)(2) pathway (Seghal Sanjay 2013)
- Chemistry, Manufacturing and Controls (CMC) requires analytical requirements and stability studies
- Active pharmaceutical ingredients data requirements for a new submission include differentiation of particle characterization, stress-studies, photostability data, impurity characterization, etc.
- Safety, Efficacy, and toxicity studies are usually required, and they can include Preclinical/toxicological data – single and repeat dose
- Carcinogenicity data
- Chronic dermal toxicological data and repeat dermal toxicological data (for transdermal delivery product)
- Carcinogenicity potential and local tolerance data
- Reproductive toxicological data, Geno-toxicological data
- Patent certification is required
- Pharmacokinetic (PK) data – minimum of single dose fasted bioequivalence (BE), single dose food-effect BE (almost always required), multiple-dose, steady-state study for modified release products is almost always required
- Labeling – may be like the reference drug, but may include unique data generated for submission, though some indications may differ from reference drug product (Seghal Sanjay 2013).
1.4 Time and cost for 505(b)(2) NDA
Table 1: Time and cost for 505(b)(2) NDA

1.5 The 505(b)(2) Process
The major elements in the process of 505(b)(2) NDA are described below
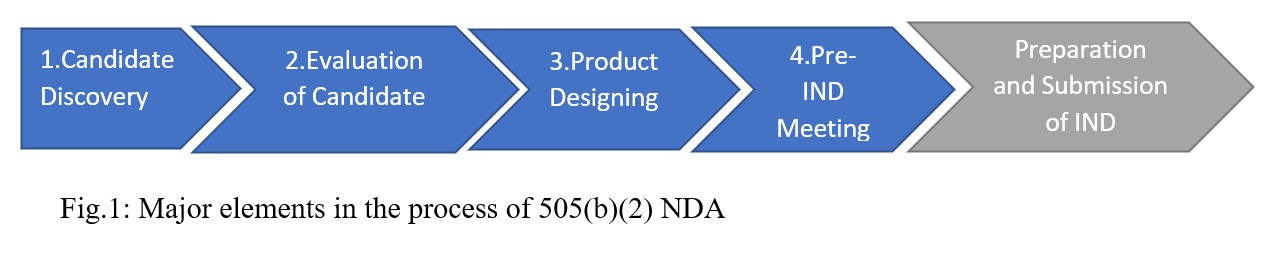
Fig.1: Major elements in the process of 505(b)(2) NDA
- Identifying the drug candidate begins the 505(b)(2) process. Potential candidate will include
- Candidate drugs with novel indications or active pharmaceutical ingredients, changes in dosage form, strength, formulation, dosing regimen or route of administration
- Candidates for new combination, either drug-drug or drug-device combinations
- Prodrugs of an approved or existing drug (CDER FDA, 1999).
Table 2: Drug types that can be identified for 505(b)(2)
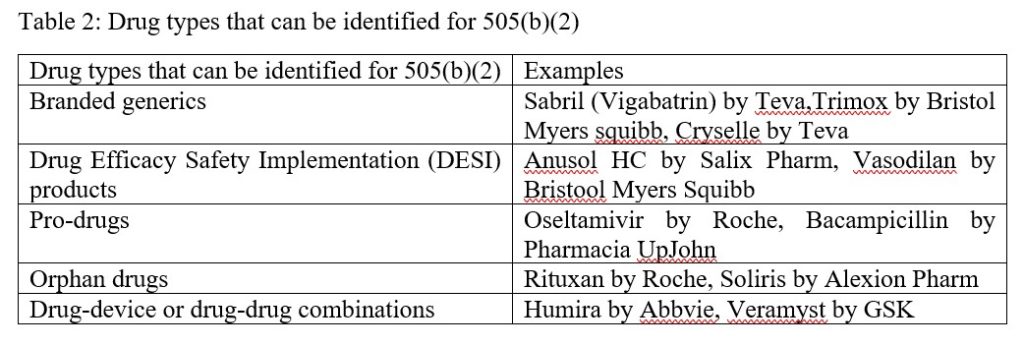
Possible types of drugs that can be identified for development through the 505(b)(2) pathway include branded generics, Drug Efficacy Safety Implementation (DESI) drugs, pro-drugs, orphan drugs, and drug-drug or drug-device combinations. It is worth noting that biological therapeutics and biosimilars are not suitable under 505(b)(2), these are registered through the BLA program (The Biologics License Application (BLA) is a request for permission to introduce, or deliver for introduction, a biologic product into interstate commerce (Biologics License Applications (BLA) Process (CBER FDA).
2. Next phase in the process is evaluating the identified drug candidate which describes the product’s feasibility. This phase measures the viability of the drug candidate such as scientific viability, medical practicality, regulatory feasibility, and commercial viability of the product of interest. It gives a cushioning foundation to planning the product’s development (CDER FDA 1999).
3. Furthermore, product designing which is the next step in the 505(b)(2) process describe ways to lower the risk of attrition or product failure by incorporating existing data into the development strategy (Maurer et al, 2021). Due to the 505(b)(2) pathway benefit of lower cost and faster approval time, some products may qualify for distinction or special treatment in cases such as
- Orphan drug exclusivity for 7 years
- New chemical entity exclusivity for 5 years
- Pediatric exclusivity, for which 6 months is added to existing exclusivity or patent.
4. Pre-Investigational New Drug (Pre-IND) meeting follows product design stage. In general, the order of steps begins with Pre-IND meeting, then formulation development (and studies if necessary), to preparation and filing Investigational New Drug (IND). The goals and strategies of pre-IND meeting are to gain FDA input on necessary data agreement with – the preclinical studies or clinical trials, the chemistry, manufacturing, and control (CMC), initial drug development plans and regulatory requirements. For the number and type of studies required, the pathway may allow for programs to conduct bridging studies that may prevent the need for some clinical or non- clinical studies traditionally required for a 505(b)(1). This also supports the idea that because 505(b)(2) development plans rely largely on existing data, clinical trials can often be started simultaneously and developed in parallel, significantly shortening the overall development timeline of the product (Strovel et al, 2016). The decision to submit an IND investigation will assume that all required sections of the application have been met.
2.0 STUDIES IN FILING FOR CHANGE IN ROUTE OF DRUG ADMINISTRATION
In 2019, Salminen and colleagues discussed that in modifying dosage forms of already existing drugs, nonclinical requirements could depend on data from previous preclinical studies of the old drug (that is, new animal/nonclinical studies may not be necessary to support safety claims). Examples of such change include reformulating an oral tablet to an oral soluble film tablet form and changing an immediate-release oral drug to and extended-release form. But the authors (Salminen et al 2019) also stated that in giving an old drug a new route of administration, preclinical tests may be necessary to show information about the local safety of the new route. Like in a case of an oral tablet which is the reference product , suppose the proposed new route is intramuscular administration, then a preclinical toxicology evaluating the local safety of the injection should be done except if it can be scientifically justified that the study is not needed (Salminen et al 2019).
2.1 Example of analyses conducted for change in route of administration
In 2017, while studying undesirable patient experiences from a specific analgesic drug, Zunhammer et al noted that changing the administration route of the drug may minimize patients’ poor view about the drug. In their study to investigate if changing the drug’s administration route could lessen such views, Zunhammer et al pooled data from 6 research results in healthy human volunteers. They employed an experimental methodology because of the ethics governing human trials, pooling a positive or negative experience data with a topical analgesic in a mock clinical study setting. Using this methodology, the treatment potency of the analgesic was studied for two days before they introduced a topical placebo drug and another placebo with oral administration on the third day. Then the efficacy of the analgesic drug was evaluated, and the potency of the treatment was defined as analgesic feedback in an experimental heat-pain model using pain intensity ratings (the visual analog scale).
From their results in 2017, participants’ treatment expectations changed over the course of the experiment. Prior to the study, all the volunteers expected similar outcomes, but these expectations began to change on the second day. The outlook of both positive and negative volunteers started to change. The positive experience group of volunteers which had an analgesic effect during exposure were high in expectations while the negative experience groups with no analgesic effect during the exposure sessions had less expectations. When the new treatment and route of administration was announced on the third day, the treatment experience expectation was notably lesser in the positive experience group than in the negative volunteers. Zunhammer et al (2017) were able to show proof that a drug’s route of administration can increase patients’ experience, hence encourage treatment compliance.
- MEDICINES FOR WHICH ADMINISTRATION ROUTES WAS CHANGED
3.1 Methotrexate Auto Injector Pen
The sponsor relied on past literature on Oral Methotrexate and Methotrexate injection to support the claim for safety and efficacy of the new administration route – subcutaneous (SC) – for the Rheumatoid Arthritis (RA) and Psoriasis indications. Prior to this, SC route was approved for Polyarticular Juvenile Idiopathic Arthritis (CDER App 205776, 2014). The sponsor submitted safety and efficacy study results for the oral, intramuscular (IM), and subcutaneous (SC) forms of methotrexate to support the filing for the auto injector pen.
For Methotrexate Intramuscular injection (MTX IM), the sponsor relied upon information from the Listed Drug, i.e., the reference product (MTX Oral). Information derived from the listed drug to support the new administration route include dosage forms and strength, indications, clinical pharmacology, safety, and toxicology. (CDER App 205776 2014).
The US FDA has advised that when relying on information from previous studies, whether of a previously approved product or from published literature, such information must be scientifically appropriate. The company applying for the new drug must present a scientific connection to support the relationship of the Listed Drug (reference) and intended product (CDER App 205776 2014).
Sometimes in changing the administration route of a drug, bridging studies are necessary to give pharmacokinetic, pharmacodynamic, or other clinical data on efficacy, safety, dosage, and dose regimen. Where bridging studies are not needed (or justifiably unavailable), pharmacokinetic information can be used alone (ICH, 1998). Bridging studies done for the methotrexate auto pen product were Bioavailability and Bioequivalent studies (BA/BE studies), a human factor (HF) study, and data on safety and efficacy of SC route for Psoriasis and RA indications.
- one primary study on bioavailability was performed to bridge the MTX autoinjector pen to the reference drug – oral MTX Product. This study compared the relative BA of the SC route to the reference drug then the results backed up the claim of the efficacy of SC dosing in RA and Psoriasis patients. Increased systemic concentration of MTX via the SC route borders the already available information on efficacy and safety with the oral reference MTX.
- a second study on BA bridged the MTX auto injector pen to the other reference product- the IM MTX injection.
- one Human Factor Study- which reviewed the effect of body weight on the new route and evaluation of the injection site were added.
- the applicant company also counted on published literature for the efficacy and safety of SC administration for the RA and Psoriasis indications (CDER App 205776 2014).
3.2 Narcan Intranasal (Naloxone Hydrochloride)
The 505(b)(2) new drug application for Narcan as filed by Adapt Pharma in July 2015 offered to modify both dosage form and route of administration. Using already approved Narcan injection (NDA 016636) as reference drug, the company created an intranasal product. This product application relied upon a relative bioavailability study in healthy population of volunteers. But because the FDA had earlier stopped the distribution of Narcan injection, the company had to rely on a generic product, International Medicinal System’s naloxone HCl injection USP pre-filled syringe (ANDA 072076) as the reference product for the relative bioavailability study. This was required to show a scientific bridge to the agency’s previous studies on Narcan. Nonclinical studies were not required for this application because it presented a lower strength (2mg naloxone hydrochloride) than the reference product (4mg naloxone hydrochloride) (Narcan Intranasal Summary Review for Regulatory Action 2017).
Although it was made for use in opioid crisis situations, intranasal Narcan was approved through priority review based on its public health importance for acute drug addiction, people who could administer the drug in an emergency situation, and patients with opioid prescriptions. And it became the first intranasal naloxone product to be approved.
3.3 Zolpidem Tartrate sublingual tablet: Immediate Release to Sublingual tablet
First approved in 1999 for the treatment of somnipathies, Edluar (zolpidem tartrate) was approved to treat sleep difficulty on a short term. Both nonclinical and clinical studies, and evaluations relevant to safety concerns were conducted for this application.
The clinical trials carried out with the reference immediate release zolpidem tartrate in support of efficacy of the new sublingual tablets were 4-5 weeks in duration, then the final formal evaluations of sleep latency were performed at the end of treatment (Edluar Drugs@FDA 2019).
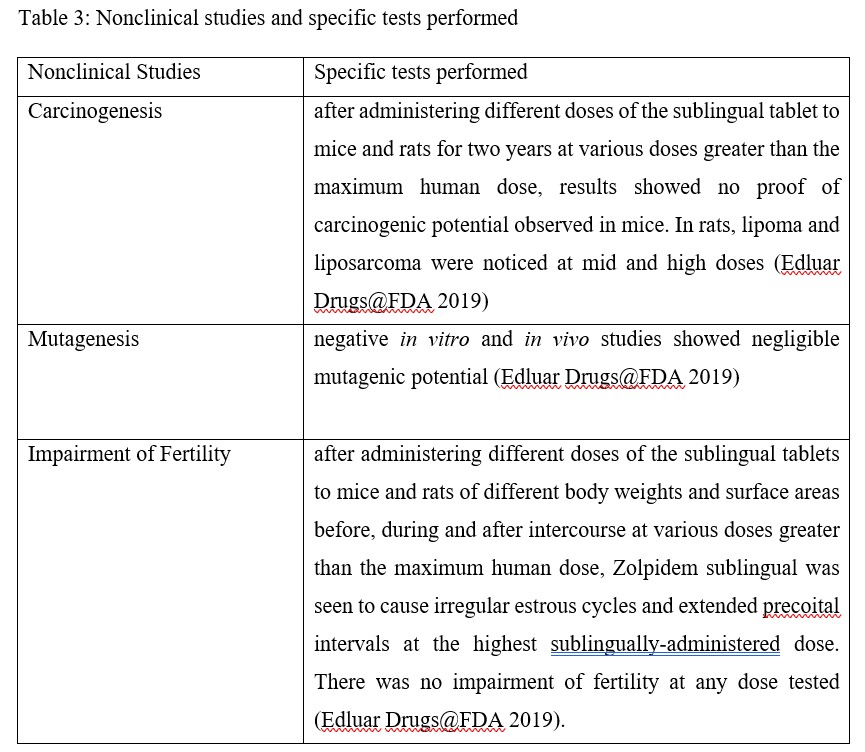
Non-clinical studies involved those for mutagenesis, carcinogenesis, and fertility impairment
Table 3: Nonclinical studies and specific tests performed
Clinical Studies
Table 4: Clinical studies and specific tests performed

Additional specific clinical studies for drugs of the sedative/hypnotic class were conducted. These include next-day residual test, rebound effects test, memory impairment test, and effects on different stages of sleep (Edluar Drugs@FDA 2019).
These clinical assessments serve as basis for the warning and precaution section of the sublingual zolpidem tartrate label. Results from both nonclinical and clinical studies of the new product give reasons for the adverse effects stated of the drug label such as CNS depressing effects, abnormal thinking or behavioural effects, and respiratory depression.
In a mild reflection, even though there are fixed guidelines for studies or data regarding new drug submission, some assessments that are drug-based or drug class-dependent must be carried out to ensure that the potential product will not cause harm to patients.
3.4 Mesalamine Extended-Release Capsule: Rectal Enema to Extended-Release Oral Capsule
PENTASA (mesalamine) for oral administration is an extended-release formulation of mesalamine, an aminosalicylate anti-inflammatory agent for gastrointestinal use (PENTASA Drugs@FDA 2021).
Clinical studies submitted for approval: two randomized, double-blind, placebo-controlled, dose-response trials of patients with active mild to moderate ulcerative colitis.
Information for the nonclinical data for this submission was not mentioned (PENTASA Drugs@FDA 2021).
CONCLUSION
The benefits of incrementally modified drugs cannot be over-emphasized. Improving drugs through change in route of administration not only enhance patients therapy adherence but also gives better health outcomes and quality of life. Changing some drugs route of administration is sure to improve safety and efficiency of healthcare professional resources, increase treatment options and prevent therapeutic escalation, while expanding cost-effectiveness and ultimately access to quality healthcare. Modification can be through drug repositioning, reformulation, and drug combination (drug/drug or drug/device or drug/service). Challenges with understanding regulatory requirements can be salvaged in a detailed pre-IND meeting, where applicants are required to demand all the necessary information that will help their product application. Because candidate drugs differ in class, origin, and even dimension, sponsors are expected to prepare to submit data showing complete safety and efficacy reports for their potential products, because only clinical and/or nonclinical studies may not be enough. There are many unexploited opportunities in this area of drug development; although a major drawback is that applicants/sponsors (especially those from small-scale companies and start-ups) are not familiar with the regulatory pathways for registering these kinds of “new drugs”. There is substantial untapped potential all over the world to modify old drugs or existing therapies to best meet the needs of patients and healthcare professionals.
REFERENCES
Abbreviated Approval Pathways for Drug Product: 505(b)(2) or ANDA? 2019 https://www.fda.gov/drugs/cder-small-business-industry-assistance-sbia/abbreviated-approval-pathways-drug-product-505b2-or-anda-september-19-2019-issue accessed 22nd Jan 2022.
Applications Covered by Section 505(b)(2) 1999 https://www.fda.gov/regulatory-information/search-fda-guidance-documents/applications-covered-section-505b2 accessed 22nd Jan 2022
Berndt ER, Cockburn IM, Grépin KA. The impact of incremental innovation in biopharmaceuticals: drug utilisation in original and supplemental indications. Pharmacoeconomics. 2006; 24 (Suppl 2): 69-86. doi: 10.2165/00019053-200624002-00008. PMID: 23389490.
Biologics License Applications (BLA) Process (CBER) https://www.fda.gov/vaccines-blood-biologics/development-approval-process-cber/biologics-license-applications-bla-process-cber accessed 12th Jan 2022) .
Cder App 205776 2014 Center For Drug Evaluation And Research Application Number: 205776orig1s000 Other Review(S) 2014 https://www.accessdata.fda.gov/drugsatfda_docs/nda/2014/205776Orig1s000OtherR.pdf accessed 8th Jan 2022)
CDER FDA Guidance for Industry Applications Covered by Section 505(b)(2) October 1992. https://www.fda.gov/media/72419/download accessed 28th April, 2022.
Edluar Drugs@FDA 2019 https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/021997s011lbl.pdf accessed 12th Jan 2022)
Globerman S. and Lybecker K. M. Chapter 2 in The Benefits of Incremental Innovation: Focus on The Pharmaceutical Industry page 23-25 Fraser Institute June 2014. https://www.fraserinstitute.org/sites/default/files/benefits-of-incremental-innovation.pdf accessed 21st April, 2022
ICH Harmonized Tripartite Guideline: Ethnic Factors in The Acceptability of Foreign Clinical Data E5(R1). 1998. Guideline for Clinical Practice. https://database.ich.org/sites/default/files/E5_R1__Guideline.pdf
Komajda M, Böhm M, Borer JS, Ford I, Tavazzi L, Pannaux M, Swedberg K. Incremental benefit of drug therapies for chronic heart failure with reduced ejection fraction: a network meta-analysis. Eur J Heart Fail. 2018 Sep;20(9):1315-1322. doi: 10.1002/ejhf.1234. Epub 2018 Jun 19. PMID: 29806165.
Maurer TS, Edwards M, Hepworth D, Verhoest P, Allerton CMN. Designing small molecules for therapeutic success: A contemporary perspective, Drug Discovery Today, 2022; 27(2): 538-546, ISSN 1359-6446, https://doi.org/10.1016/j.drudis.2021.09.017
Mithilesh Kumar Jha. Modified Release Formulations to Achieve the Quality Target Product Profile (Qtpp) IJPSR, 2012; 3(8): 2376-2386 https://www.researchgate.net/publication/265946962_MODIFIED_RELEASE_FORMULATIONS_TO_ACHIEVE_THE_QUALITY_TARGET_PRODUCT_PROFILE_QTPP accessed 21st April, 2022
Narcan Intranasal Summary Review for Regulatory Action https://www.accessdata.fda.gov/drugsatfda_docs/summary_review/2017/208411s001SumR.pdf accessed 23rd Jan 2022)
Lee Hye-seon. Domestic pharmaceuticals active to develop drugs by changing combinations or administration routes. Korea Biomedical Review 2017 http://www.koreabiomed.com/news/articleView.html?idxno=588#:~:text=The%20incrementally%20modified%20drugs%20refer,mixes%20two%20or%20more%20compounds. Accessed 20th Jan 2022.
PENTASA Drugs@FDA 2021 https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/020049s035lbl.pdf accessed 12th Jan 2022
Salminen W.F., Wiles M.E., Ruth E.S. 2019 Streamlining nonclinical drug development using the FDA 505(b)(2) new drug application regulatory pathway. Drug Discovery Today Volume 24.
Seghal Sanjay 2013 https://www.pharmacy.umaryland.edu/media/SOP/wwwpharmacyumarylandedu/centers/cersievents/SehgalNotes.pdf accessed 22nd Jan 2022.
Schuhmacher, A., Gassmann, O. & Hinder, M. Changing R&D models in research-based pharmaceutical companies. J Transl Med 2016; 14: 105. https://doi.org/10.1186/s12967-016-0838-4
Strovel J, Sittampalam S, Coussens NP, et al. Early Drug Discovery and Development Guidelines: For Academic Researchers, Collaborators, and Start-up Companies. 2012 May 1 [Updated 2016 Jul 1]. In: Markossian S, Grossman A, Brimacombe K, et al., editors. Assay Guidance Manual [Internet]. Bethesda (MD): Eli Lilly & Company and the National Center for Advancing Translational Sciences; 2004-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK92015/ accessed 28th April, 2022.
Zunhammer M., Ploner M., Engelbrecht C., Bock J., Kessner S., Bingel U. 2017. The effects of treatment failure generalize across different routes of drug administration. Science Translational Medicine Vol 9, Iss 393.