1.0 INTRODUCTION
Tablets are the unit solid dosage forms meant for oral use and are manufactured by using tablet compression machines, by compressing the active pharmaceutical ingredient with or without other excipients [1]. An excipient is a natural or synthetic substance formulated alongside the active ingredient of a medication, included for the purpose of bulking-up formulations that contain potent active ingredients (thus often referred to as “bulking agents”, “fillers” or “diluents”) or to confer a therapeutic enhancement on the active ingredient in the final dosage form, such as facilitating drug absorption or solubility [2]. Excipient research and development is an area in Pharmaceutics that has gained popularity, especially with the renewed desire to shift from the conventional dosage forms to the modified release dosage forms which impart some characteristics to the release profile of the active drug. Drug modification involves altering the release profile, site of absorption or the rate of absorption of a drug in plasma in order to either reduce the frequency of drug administration, improve patient compliance and/or to reduce the side effects of the drug [3].
1.1 Modified Release Delivery Systems
Modified release delivery systems are defined by the USP as those whose drug release characteristics of time course and/or location are chosen to accomplish therapeutic or convenience objectives not offered by conventional dosage forms and may be classified into five categories:
- Delayed Release: Here, the drug release is delayed after administration for some period of time to evade deleterious environmental conditions such as gastrointestinal pH.
- Repeat action: This contains two or more doses of the drug, one for immediate release and subsequent doses are delayed.
- Receptor targeting: These systems of delivery refers to targeting of a drug directly to a certain biological location. In this case the target is the particular receptor for a drug within an organ or tissue.
- Site specific targeting: These systems refer to targeting of a drug directly to a certain biological location, the target being adjacent to or in the diseased organ or tissue.
- Sustained release: Here, the system provides an initial release of drug sufficient to provide a therapeutic dose soon after administration, and then a gradual release of drug over an extended period of time. It can also provide some control of drug release in the body, whether this be of a temporal or spatial nature, or both. Or in other words, the system is successful at maintaining constant drug levels in the target tissue or cells.
- i) Controlled Release Systems: These systems include any drug delivery system that release drug at a constant rate and provide plasma concentrations that remain invariant with time.
- ii) Extended Release Systems: These are pharmaceutical dosage forms that release the drug slower than normal manner at predetermined rate, and necessarily reduce the dosage frequency by two folds.
Interestingly, the USP considers that the terms ‘controlled release’, ‘prolonged release’ and ‘sustained release’ are interchangeable with ‘extended release’ but from a biopharmaceutical point of view, this is not strictly a concern [4].
To achieve sustained release of drugs, two design systems; Reservoir system and Monolithic (Matrix) systems can be utilized. In the reservoir system, the drug first partitions into the membrane from the reservoir and then diffuses to the other side of the membrane, where it is taken up by the receiving medium. As the reservoir becomes saturated, a constant concentration gradient of the drug is sustained in the membrane, the rate of drug flux is constant, and zero order release is attained. Finally, the drug concentration in the reservoir drops below saturation, and the gradient across the membrane and release rate both deteriorate. Here, the purpose of the membrane is to mediate diffusion of the drug. Because of their simplicity of mechanism and their ability to produce zero order release, reservoir systems would appear to be greatly advantageous. However, reservoir systems can be challenging to fabricate reliably as minor defects and cracks in the membrane can lead to dose dumping. These problems however are avoided in matrix systems in which the drug is loaded directly into a polymer, which now acts as both a storage medium and a mediator of diffusion. The drug is typically loaded homogenously into the matrix device, and the drug release is controlled by its diffusion through the matrix material or through aqueous pores. Matrix devices typically exhibit an initial burst release from the surface but, as time passes, the release rate decreases because the drug that is deeper inside the matrix must first diffuse to the surface, and since it has to travel farther, the quadratic relation between distance and time becomes important. This effect which occurs in planar monoliths becomes even more prominent with cylinders or spheres, as the amount of drug available decreases with distance from the surface [5].
1.2 Cross-linking of Purified Acacia seyal gum
Cross-linking involves re-enforcing bonds with chemical bridges between molecules or polymer chains. A cross-link is a bond that links one polymer chain to another, which affects the physical characteristics of polymers to include particle properties, hydration and water sorption capacities as a few of such properties which may lead to the cross-linked gum becoming more resistant to high temperatures, high shear and an improved viscosity. This is seen in the ionic gelation method adapted by Akila et al., (2020), to cross-link Acacia seyal gum powder with freshly prepared 1M Calcium chloride solution[6].
Suitability of Drug for Formulation as Sustained-Release Dosage Form
Suitability of the drug has to be considered before development of sustained-release dosage form. The following must be taken into consideration;
- Solubility of the drug in aqueous media and the intestinal permeability of the drug: drugs with high solubility and permeability are most suited for extended-release delivery as drug release from the dosage forms can be the rate-limiting step in the process (low solubility; <1 mg/ml). Drugs with low permeability (<0.5×10-6 mms-1) are unlikely to be suited for extended-release as they are already rate-limited in their absorption.
- Elimination from blood stream: the most suitable drugs may have relatively short half-lives of 4 – 6 h.
- Dose: to limit the size of the dosage form, the potency of the drug in the modified-release form can be critical. Up to 1000 mg potency tablets are available but this is only achieved by using very large tablets, which may not always be acceptable for some patient populations.
1.3 Salbutamol
Fig. 1: Salbutamol chemical structure
Salbutamol sulphate is a white or almost white odourless powder. It is soluble in four parts of water and chemically described as 1-(4-hydroxy-3-hydoxymethylphenyl)-2-(tert-butyl amino) ethanol sulphate. A β-2 adrenergic agent with bronchodilatory effect and is useful in the treatment of asthma and management of Chronic Obstructive Pulmonary Disease (COPD. Salbutamol sulphate must be dosed three to four times daily to maintain its bronchodilatation effect due to the short half-life (2.7 – 5 h). Therefore, to reduce the frequency of administration and to improve patient compliance, a once or twice daily sustained release formulation of Salbutamol sulphate is desirable. The drug is freely soluble in water, and hence judicious selection of release retarding excipients is necessary to achieve a constant in–vivo input rate of the drug .
2.0 METHODS
2.1 Materials
Purified Acacia seyal gum powder (EG), Cross-linked Acacia seyal gum powder (CG), Hydroxypropyl methylcellulose (JRS PHARMA, HPMC E15, Zacapu, Mexico), Lactose anhydrous (BDH, England), Magnesium stearate (BDH, England), Talc (BDH, England), Distilled water, Deionized water, Salbutamol sulphate (Avrishtava PVT, India). All reagents used are of analytical grade.
EG and CG were obtained and characterized according to the methods described and reported by James, Isah and Olowosulu., (2020), referenced as Akila et al., (2020) [6].
2.2 Preparation of Salbutamol Sulphate Sustained-Release Tablet Formulations
The formula for preparation of sustained release tablets of salbutamol sulphate, using varying concentrations (30 % w/w, 40 % w/w and 50 % w/w) of the different polymers as matrix former is as shown in Table 1.
Table 1: Formula for the Formulation of Sustained-Release Salbutamol Sulphate Tablets.
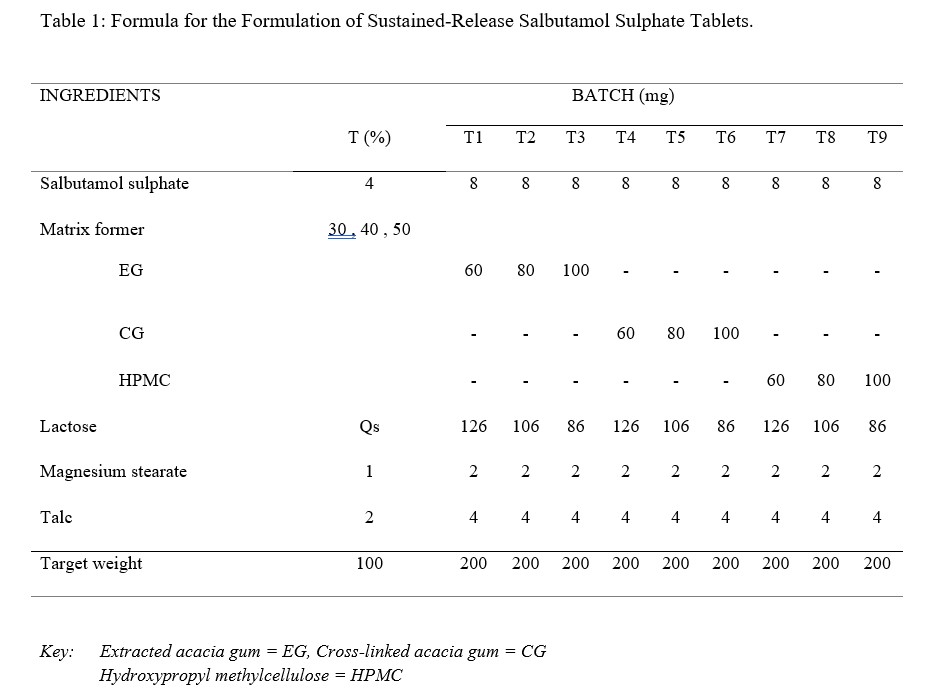
A total of 70 tablets per batch were made. The concentration of Salbutamol sulphate was kept constant for all batches and the target weight of each tablet was 200mg. Salbutamol sulphate and all excipients except talc and magnesium stearate were weighed accurately, and blended in a mortar with the help of a pestle for 5-10 min. The required amount of talc and magnesium stearate were added and further mixing was done for 4-5 min. The powder blends were then compressed using Erweka EK AR 400 Eccentric Tablet Press (Germany), with the diameter of the punch and die as 8 mm and a compression pressure of 6.5 – 7 kgN. The compressed tablets were of convex round shape.
2.3 Evaluation of Tablets
All batches of prepared tablets were evaluated for various tableting parameters as follows:
2.3.1 Weight Variation
For uniformity of weight, twenty tablets from each batch of formulations were selected at random and their individual weights determined by using an electronic balance. The average weight and standard deviation of the tablets were then calculated [4].
2.3.2 Diameter
Ten tablets from each batch of the formulations were selected randomly and their diameters measured thrice, using a digital caliper. The average value of the diameter was then calculated for each batch.
2.3.3 Thickness
Ten tablets from each batch of the formulations were selected randomly and their thickness measured thrice using a digital caliper. The average value of thickness was calculated for each batch.
2.3.4 Friability
Friability test was carried out using Roche Friabilator. Five (5) tablets were weighed (Wo) and subjected to combined effect of attrition and shock by utilizing a plastic chamber that revolves at 25 rpm, dropping the tablets at a distance of 6 inch with each revolution, operated for 100 revolutions. The tablets where then dusted, reweighed (W) and the percentage friability calculated using equation 1.

2.3.5 Crushing Strength
The crushing strength (KgF) of the prepared tablets were determined by using a monsanto hardness tester. The crushing strength tests was performed for each batch of prepared tablets in triplicate. The average crushing strengths and standard deviations were then determined.
2.3.6 Tensile strength
The equation below described by Fell and Newton in 1970 was used to calculate the tablet tensile strength for the tablets prepared [7].

where T = Tensile strength; F = Crushing strength; d = diameter; t = thickness
2.3.7 Disintegration Test
This test was carried out using the Coupley DTG 4000 disintegration tester. Three tablets per batch were placed in the cylindrical tubes and set into up and down motion, mimicking the GI movement. The disintegration time was recorded as the time taken for the last palpable fragment of the tablet to leave the mesh in the cylinder [4].
2.3.8 Uniformity of Drug Content
Five tablets were weighed individually and the mean weight determined. The tablets were then powdered in a porcelain mortar, and 200 mg equivalent of the tablet triturate was taken in a 100 ml volumetric flask. The volume was made up to mark with simulated gastric fluid (0.1N HCl) to give Stock solution (I). A 10 ml volume was transferred to a precalibrated 100 ml volumetric flask and diluted up to the 100 ml mark with 0.1N HCl. The solution was filtered through a filter paper and its absorbance was measured spectrophotometrically (Shimadzu, Japan), with 0.1N HCl as blank, at 277nm [8].
2.3.9 In–vitro Drug Release Studies
In-vitro release of Salbutamol sulphate from sustained release tablets was determined using USP type II dissolution apparatus in 900 mL of 0.1N HCl for 2 h and phosphate buffer (pH 6.8) for 6 h at constant temperature of 37±0.5 °C and 100 rpm. Aliquots (5 ml) of the solutions were withdrawn from the dissolution apparatus at different time intervals and replaced with fresh dissolution medium to maintain the sink condition. These aliquots were then filtered and the absorbance of these solutions measured using a double beam ultra-violet spectrophotometer (Shimadzu, Japan) at 277 nm against fresh 0.1N HCl (pH 1.0) and phosphate buffer (pH 6.8) solutions as blanks, based on the dissolution media used for a particular sample. The percent drug release and cumulative percent drug released were then calculated using the data obtained from the calibration plot of salbutamol sulphate [8].
2.3.10 Release Kinetics
Data obtained from in–vitro release studies were fitted into various kinetic equations to find out the mechanism of drug release from the polymers. The following kinetic models were used:

Determining the correlation coefficient will help assess fitness of the data into various kinetic models. The rate constants for the respective models were obtained from the slopes of the graphs plotted.
2.3.11 Statistical Analysis
The results obtained from the tests that required analysis were analyzed as mean ± standard deviation (±SD), using Microsoft Excel (Office 2013 version) and the release profiles were analyzed using Univariate analysis of variance (IBM SPSS Statistics v20) at significance level of p ≤ 0.05.
3.0 RESULTS AND DISCUSSIONS
3.1 Tablet Properties
The ease, simplicity and requirement for absence of moisture due to the formulation components informed the choice of direct compression method for this research. Weight variation is mainly due to poor flow of material into the die, which leads to improper filling of the die. From table 2 below, the weight variation observed for all batches ranged between 0.201±0.004 g to 0.204±0.004 g and this is within the USP and BP recommendation which allows a 7.5 % variation in weight for tablets with weight between 131 mg – 325 mg.
Table 2: Properties of Sustained-Release Salbutamol Sulphate Tablets Formulated
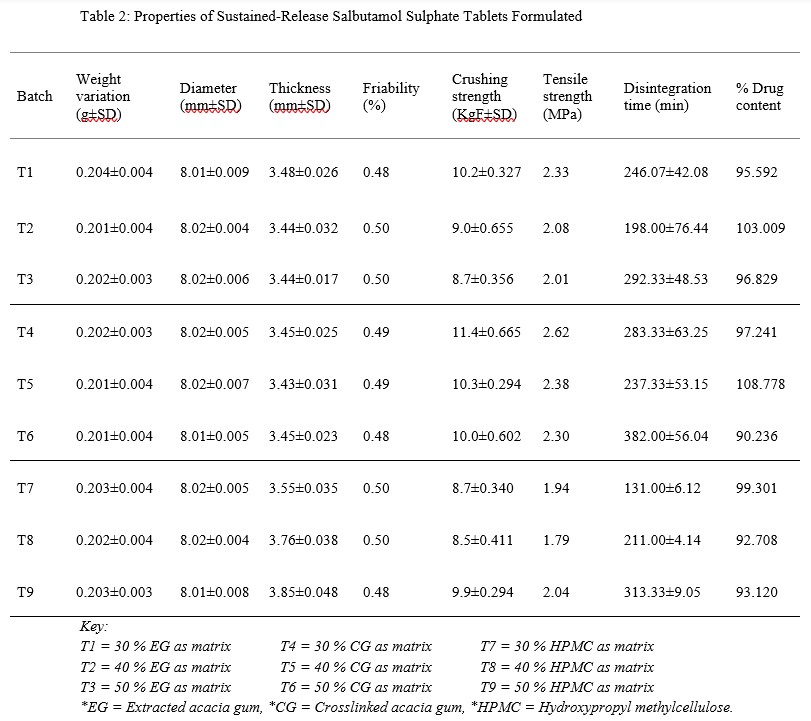
Tablet thickness, diameter and crushing strength are widely used by tablet manufacturers to check the quality of a batch and are important quality control tests for tablet packaging. The thickness of the tablet can affect the therapeutic effectiveness of tablets and very thick tablets affect packaging either in blister or plastic container [9]. Tablet thickness is usually controlled to minimize appearance problems to assure that the tablet fit into the container and gives assurance that they can be accurately counted by the filling equipment as some filling equipment depend on the uniform thickness of the tablets as a counting mechanism. B.P and U.S.P standards suggest that all of the percentage average deviation value must not exceed ±5 % for tablets with diameter less than 12.5 mm. Tablets must have a certain degree of strength to be able to withstand and overcome the rigors of mechanical shock so that they do not break easily. They should be able to also withstand reasonable abrasion and friction. Adequate tablet hardness and resistance to powdering and friability are necessary requisites for consumer acceptance. It also may influence disintegration and affect the dissolution rate. It may be especially important to carefully monitor crushing strength for drug products that possess real or potential bioavailability problems or are sensitive to altered dissolution – release profiles as a function of the compressive force employed. The friabilities of all the batches were within the recommended limit of < 1 %, as they ranged between 0.48 to 0.50 %. However, this may have been caused by the relatively high concentration of the binders used as matrix former. Oral tablets normally have a crushing strength of 4 – 8 KgF. However, hypodermic and chewable tablets are much softer (3 KgF) and some sustained release tablets are much harder (10 – 20 KgF). The higher crushing strength values obtained with batches containing CG and EG as matrix formers compared to those formulated with HPMC might be due to the fact that CG and EG are less dense than HPMC [6], thereby allowing effective pressure transmission during compression and resulting in the well knitted particles observed with CG. Disintegration is considered to be that state whereby no residue remains on the screen as a soft mass having palpable firm core, except fragments of insoluble coating [10]. The disintegration time observed had a range between 131.00±6.12 min to 382.00±56.04 min. Disintegration test is however not really applicable to sustained release systems as they are made to either swell or erode gently to release the active agent. The prolonged disintegration (in this case erosion) time observed can be a pointer as to the fact that the tablets formulated are not immediate release tablets but modified for a longer delivery of the active ingredient (Salbutamol Sulphate). Generally, the higher the concentration of the matrix, the longer it takes for the tablet to erode. Though there are some exceptions to this norm, the variations in tensile strength, which may have resulted from the variation in compression pressure, may be a cause of the variation observed with the disintegration time. The USP convention, official revision bulleting of August 1, 2008 suggests for uncoated tablets, a disintegration time of 15 to 30 min. However, there are exceptions for delayed-release and coated tablets that are formulated for timed release of their active ingredients, requiring over 1 h before disintegrating. The uniformity of drug content is an important quality evaluation test for tablets with potent drug contents, which are administered in low doses, the excipients forming the greater part of the tablet weight [11]. The USP 25 suggests a range of 90 – 110 % drug content for oral tablet formulations and from the results obtained from this study, it can be seen that all the batches fell within the required standard.
3.2 In-vitro Release Studies of Salbutamol Sulphate from the Sustained Release Tablets
The dissolution profiles shown in figure 1 reveal that the release in pH 1.0 was higher than the release in pH 6.8.
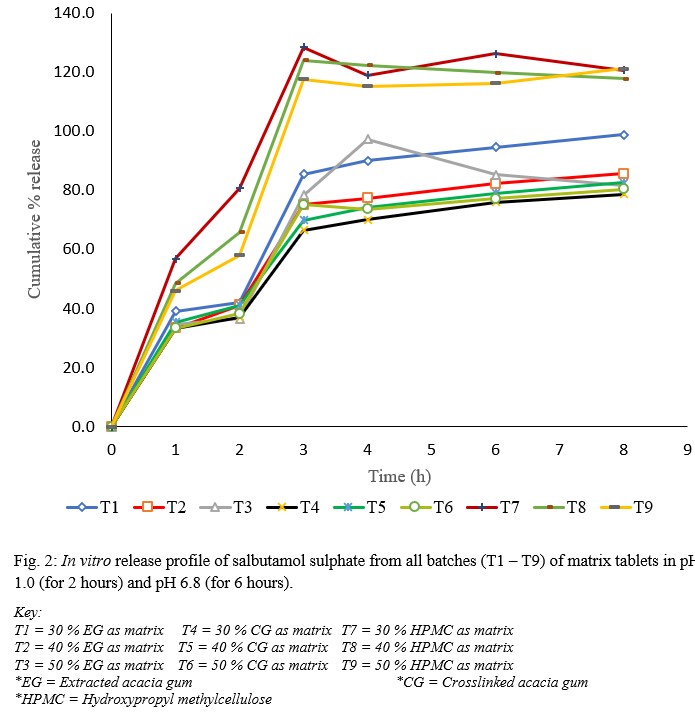
Fig. 2: In vitro release profile of salbutamol sulphate from all batches (T1 – T9) of matrix tablets in pH 1.0 (for 2 hours) and pH 6.8 (for 6 hours).
This was thought to be as a result of dissolution of the drug from the surface and near the surface of the matrix, which occurred while the polymer was undergoing hydration to form the gel layer. This result is in line with report by Nep et al., (2016) [12] who studied the release of theophylline from Sesamum gum and suggested that the drug diffusion front movement was the main parameter affecting drug release rate. Also worthy of note from the dissolution studies is the fact that formulations made with CG as polymer matrix exhibited better salbutamol-sustaining ability when compared with the other polymers (EG and HPMC). This goes to mean that the cross-linking procedure may have conferred on the gum some ability(ies) that makes it a better polymer in sustaining the release of the salbutamol from its matrix. Some of the abilities that may have been conferred on the gum on crosslinking are reduced solubility, higher molecular weight and improved viscosity. From the physicochemical characterizations reported by Akila et al., (2020) [6], it was shown that CG was only sparingly soluble in water, has a better hydration capacity, swelling ratio and swelling rate as compared to EG. Once the tablets were in contact with the dissolution medium, the penetrating water causes CG to swell rapidly. The thickness and decreased porosity of the diffusion layer thus formed inhibits the passage of the drug molecules and this may have been the reason for the better drug release retarding property of CG, as water could not penetrate easily to dissolve the salbutamol in the formulation [13]. Drug release from sustained release formulations is influenced by polymer molecular weight probably because at higher molecular weights, the polymer was entangled and the effective molecular diffusion area reduced. It has been reported that there is a linear correlation between the proportion of polymer in the tablets and the extension of drug retardation. This is because the concentration of the polymer affects the gel strength, resulting in stronger diffusional layer that is resistant to diffusion or erosion, ultimately slowing down drug release. On one hand, lactose (used as diluent/filler/bulking agent) dissolves quickly, leaving fluid filled pores and channels that allow quicker medium penetration and drug release; on the other hand, the polymer swells and the resultant gel blocks the pathway of the medium and the drug, thus slows down medium penetration and drug release [13, 14]. In this study, the lower polymer proportions exhibited better sustaining capacity as observed with T4 (30 % CG proportion), which sustained better than T5 (40 % CG proportion) and T6 (50 % CG proportion); T2 (40 % EG proportion), which sustained better than T1 (30 % EG proportion) and T3 (50 % EG proportion). The HPMC matrices were however different, with T9 (50 % HPMC proportion) sustaining better than T8 (40 % HPMC proportion) and T7 (30 % HPMC proportion). A possible reason for this may be as a result of the crushing and tensile strengths of the tablet formulation. T4 from this study showed an exceptional crushing strength of 11.4 KgF and a tensile strength of 2.62 MPa. Nep et al., (2016) reported that harder compacts result from highly compactible polymers and this leads to higher crushing strength, probably due to increased number of inter-particulate hydrogen bonds during compaction. The cross-linking step may have conferred this property to CG as formulations using this polymer showed higher crushing strength values compared to EG and HPMC. The time for 50 % drug release (T50) was longer for CG matrices compared to those of EG and HPMC matrices, indicating that it takes longer to release 50 % of the drug from CG matrices. The theoretical release profile calculation is important to evaluate the formulation with respect to release rates and to ascertain whether it releases the drug in a predetermined manner [15]. It is expected that the developed formulations should have a theoretical drug release profile of about 67 % at the 8th h when using a target time of 12 h. From the result of this study, T4 is shown to have a cumulative percent release of about 79 % at the 8th h, and this is the closest any of the formulations got to the theoretical release. It can thus be concluded that T4 sustained the release of salbutamol more than any of the other formulations made so that the release is for a long time and is thus more bioavailability. For these reasons, T4 was considered the optimum formulation of this study. HPMC has been used as standard in many sustained-release studies, with successful results [12,15,16].
3.3 Release Kinetics
Based on the R2 values from the release kinetic studies presented in Table 3, the release of salbutamol from the matrices followed predominantly Higuchi model. This describes drug release that is largely governed by diffusion through water-filled pores in the matrices. Only T1 followed a predominantly First Order release model, which can be used to describe the drug dissolved in pharmaceutical dosage forms like those containing water-soluble drugs in porous material [17]. The n-values, when the dissolution data were fitted into Korsmeyer-Peppas model, except for T7 which showed Fickian diffusion, all showed values that suggest anomalous or non-Fickian diffusion. This is expected since the drug release involved a combination of both diffusion and erosion-controlled rate release [12,16].
Table 3: Release kinetics and mechanism of salbutamol sulphate transport
![]()
3.4 Statistical Analysis
A univariate analysis of variance was carried out and the effect of the polymers (materials) and their proportion of use, on the release of salbutamol from the matrix tablets formulated were examined. The descriptive statistics observed that T4 has the minimum mean release of salbutamol, while T9 has the maximum mean release of salbutamol. With this, T4 can thus be selected as the better formulation to sustain the release of salbutamol sulphate from the matrix tablets formulated. The tests of between-subjects effects showed a statistically significant difference (p = 0.000) in the release profile of the three (3) different polymers used as matrix former at < 0.05 level of significance. This implies that there is enough evidence to reject the null hypothesis which states that; Cross-linking Acacia seyal gum with calcium chloride will not impart sustained-release properties to salbutamol sulphate tablets prepared by direct compression method. We therefore concluded that; Cross-linking Acacia seyal gum with calcium chloride will impart sustained-release properties to salbutamol sulphate tablets prepared by direct compression method. We can conclude that there is difference in the mean release of salbutamol sulphate amongst the different polymers used as matrix formers in this study. The effect of the proportion of use of the various polymer materials used as matrix formers in the release of salbutamol sulphate from the tablet formulations was also examined and at < 0.05 level of significance, the p-value of 0.534 obtained shows there is no statistically significant difference in their mean. This implies that there is not enough evidence to say that the proportion of use of the polymer material used as matrix former affects the release of the salbutamol sulphate from the tablets formulated. Since there was mean difference in the release profile amongst the polymers used as matrix former, there was thus need to compare these materials using the post hoc test. The Turkey’s test was used to compare EG (material 1), CG (material 2) and HPMC (material 3). By comparing EG with CG at 0.05 level of significance, a mean difference of 6.86279 and a significance of 0.26 was observed. It can thus be said that EG has higher mean release of salbutamol sulphate than CG. Comparing HPMC with EG at 0.05 level of significance, the mean difference of 11.49004 and a significance of 0.000 was observed. It can thus be said that HPMC has higher mean release of salbutamol sulphate than EG. Comparing HPMC and CG at a significance level of 0.05, a mean difference of 18.35283 and a significance of 0.000 were observed and this implies that HPMC has higher mean release of salbutamol sulphate than CG. On a general note, CG has the better drug release sustaining property as seen from its lower mean difference compared to EG and HPMC, but the proportion of use does not show statistical significance in this study.
4.0 CONCLUSION
Based on the results obtained and the statistical analysis done, the Acacia seyal gum cross-links produced and characterized as reported by Akila et al., (2020), can be used as a matrix former, at 30 % concentration to formulate sustained-release salbutamol sulphate tablets, by direct compression method. This will help to reduce the dosing frequency of salbutamol sulphate to twice daily, as each tablet contains 8 mg, and will go a long way in improving patient’s compliance/convenience by reducing pill burden, reducing incidences of nocturnal and early morning asthmatic attacks and also providing an economical drug delivery system for management of chronic obstructive pulmonary disease.
ACKNOWLEDGEMENTS
My profound gratitude goes to my supervisory team from the Department of Pharmaceutics and Pharmaceutical Microbiology, Ahmadu Bello University, Zaria for their mentorship. Also to the Department of Pharmaceutics and Pharmaceutical Technology, Gombe State University, Gombe, for their support. Finally to the Department of Pharmaceutical Technology, University of Jos, for allowing me access to their laboratory.
CONFLICT OF INTERESTS
There are no conflicts of interest.
REFERENCES
- Tejaswi SU, Preeti G. A Brief Overview on Tablet and It’s Types. Journal of Advancement in Pharmacology, 2020; I(1): 21-31.
- Annammadevi G, Ganesh SM, Himabindu K. A Compendere of Excipients: A Review. High Technology Letters, 2021; 27(11): 592-607.
- Karunapriya Chitra, Srinath N, Rama Devi Bhimavarapu, N. Gowthami, Haritha Meda, Dhavani Kanikanti & Manasa Anne (2012): Development And In Vitro Evaluation Of Sustained Release Matrix Tablets Of Salbutamol Sulphate Using Hydrophilic And Hydrophobic Polymers. Bulletin of Pharmaceutical Research, 2(3): 112-117.
- Alderborn G. Aulton’s Pharmaceutics, The design and manufacture of medicines (4th ed.). (M. E. Aulton, & K. M. Taylor, Eds.) Leicester, UK: Churchill Livingstone Elsevier. 2013
- Siegel R, Rathbone M. Overview of Controlled Release Mechanisms. In Advances in Delivery Science and Technology, Fundamentals and Applications of Controlled Release Drug Delivery. New York, United States of America: Springer Science & Business Media. 2012: pp. 19-41
- James AB, Isah AB, Olowosulu AK. Characterization of Purified and Cross-linked Acacia seyal Gum. Journal of Pharmaceutical Development and Industrial Pharmacy, 2020; 2(1): 11-23.
- Fell JT, Newton JM. Determination of Tablet Strength by the Diametral Compression Test. Journal of Pharmaceutical Sciences, 1970; 59(5): 688-691.
- Moin A, Shivakumar HG. Formulation of Sustained-Release Diltiazem Matrix Tablets Using Hydrophilic Gum Blends. Tropical Journal of Pharmaceutical Research, 2010; 9(3): 283-291.
- San Jose State University. (n.d.). Find study resources/Tablet thickness. Retrieved April 18, 2022, from San Jose State University Web site: https://www.coursehero.com/file/107632502/TABLET-THICKNESSpdf/
- United States Pharmacopeia. Disintegration. 2006. Retrieved September 16, 2016, from General Chapters <701>: http://www.pharmacopeia.cn/v29240/usp29nf24s0_c701h.html
- Alderborn G. Tablets and compaction. In: Aulton ME, Taylor KM, editors. Aulton’s Pharmaceutics, the design and manufacture of medicines. 3rd Edition. Leicester, UK: Churchill Livingstone Elsevier; 2007: p. 665-669 and 208-210.
- Nep E, AsareAddo K, Ngwuluka N, Conway BR. Evaluation of sesamum gum as an excipient in matrix tablets. British Journal of Pharmacy, 2016; 1 (1): 74-87. ISSN 20588356
- Siriporn O, Kiattisak S, Yanee P, Helmut V. Factors Influencing Drug Dissolution Characteristic from Hydrophilic Polymer Matrix Tablet. Scientia Pharmaceutica, 2007; 75(4): 147-164.
- Caccavo D, Cascone S, Lamberti G, AngelaBarba A, Larsson A. Swellable Hydrogel-based Systems for Controlled Drug Delivery. In (Ed.), Smart Drug Delivery System. 2016. IntechOpen. https://doi.org/10.5772/61792
- Purushothaman M, Ramaiyan D, Subash V, Vijaya RJ. Development of sustained release matrix tablets of salbutamol sulphate using different polymers. International Journal of Advanced Pharmaceutics, 2012; 2(1): 5-8.
- Mahmud HS, Vercruysse, J, Remon J. In vitro and in vivo assessment of Adansonia digitata mucilage as a matrix former in modified release tablets of metoprolol tartrate. Journal of Drug Delivery Science and Technology, 2019; 54: 101227.
- Shalkh Hk, Kshirsagar RV, Patil SG. Mathematical models for drug release characterization: a review. World Journal of Pharmacy and Pharmaceutical Sciences, 2015; 4(4): 324-338. ISSN: 2278-4357.